genellik
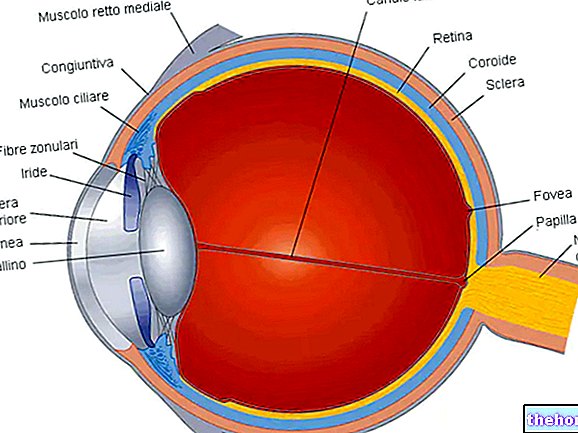
Retinoblastom (Rb), retina hücrelerinden gelişen kötü huylu bir göz tümörüdür. Bu kanser her yaşta ortaya çıkabilir, ancak başlangıç en çok beş yaşından önce bebeklik döneminde görülür.

Çocukluk çağı kanseri agresiftir: Retinoblastom lenf düğümlerine, kemiklere veya kemik iliğine yayılabilir. Nadiren merkezi sinir sistemini (beyin ve omurilik) tutar.
Retinoblastomlu çocukların yaklaşık %90'ı, teşhisin erken konulması ve kanser yayılmadan tedaviye başlanması koşuluyla olumlu bir prognoza (tedavi olasılığı) sahiptir. Mümkün olduğunda, tıbbi müdahalenin amacı hastanın görüşünü korumaktır.
nedenler
Tümör başlangıcına yol açan olaylar dizisi karmaşıktır.Bu, retinadaki hücreler kromozom 13'ün (13q14) q14 bandında yer alan RB1 tümör baskılayıcı genini içeren bir mutasyon (veya delesyon) geliştirdiğinde başlar.
Her hücre normalde iki RB1 genine sahiptir.:
- Genin en az bir kopyası doğru çalışıyorsa, retinoblastom ortaya çıkmaz (ancak risk artar);
- Genin her iki kopyası da mutasyona uğradığında veya eksik olduğunda, kontrolsüz hücre çoğalması meydana gelir.
Çoğu durumda, RB1 geninde (sporadik retinoblastom) değişiklikleri tam olarak neyin tetiklediği açık değildir; bunlar, örneğin üreme ve hücre bölünmesi sırasında meydana gelen rastgele genetik hatalardan kaynaklanabilir. Bununla birlikte, retinoblastomun altında yatan genetik anormalliklerin, otozomal dominant kalıtım paterni ile ebeveynlerden çocuklara da geçebileceği bilinmektedir. Bu, eğer bir ebeveyn mutasyona uğramış (baskın) bir gen taşıyorsa, her çocuğun onu kalıtım yoluyla alma şansı %50 ve normal bir genetik yapıya sahip olma (çekinik genler) için %50 şansa sahip olacağı anlamına gelir.
- Ara sıra bir hücre, RB1 geninin tek normal kopyasını etkisiz hale getirir (bir kopya zaten mutasyona uğramıştır);
- RB1'in iki kopyasının kaybı, "retinanın aşırı çoğalmasına" yol açar.
- Ara sıra bir hücre, normal RB1 genlerinden birini etkisiz hale getirir;
- RB1 geninin ikinci kopyası etkisiz hale getirilir;
- RB1'in iki kopyasının kaybı, retinoblastoma yol açan aşırı hücre proliferasyonunu indükler.
Genetik ve moleküler özellikler
- Retinoblastom, bir "genetik anormallik (kromozom 13'ün q14 bandının silinmesi veya mutasyonu) ile doğrudan ilişkili olan ilk tümördü.
- RB1, hücre döngüsünde önemli bir rol oynayan pRb proteinini kodlar: S fazı genlerinin (G1 → † "S) transkripsiyon kontrolüne katıldığı için DNA replikasyonuna ve hücre döngüsü ilerlemesine izin verir.
- Retinoblastoma ek olarak, RB1 geni mesane, meme ve akciğer kanserlerinde inaktive edilir.
kalıtsal retinoblastom
Kalıtsal retinoblastomlu çocuklar, hastalığı sporadik vakalardan daha erken yaşta geliştirme eğilimindedir. Ayrıca, RB1 genindeki anormallik konjenital olduğundan (yani doğumdan itibaren mevcut olduğundan) ve her ikisi de dahil olmak üzere vücuttaki tüm hücreleri (germline mutasyonu olarak bilinir) etkilediğinden, bu çocuklar diğer oküler olmayan kanserler için yüksek risk altındadır. retinalar: Bu nedenle, kalıtsal formu olan çocuklarda genellikle tek göz yerine iki taraflı retinoblastom bulunur.
Belirtiler
Daha fazla bilgi için: Retinoblastom Belirtileri
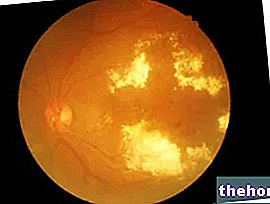
Retinoblastomun en yaygın ve bariz belirtisi, bir ışık huzmesi (lökokori veya amaurotik kedi refleksi) çarptığında grimsi beyaz bir yansıma sunan öğrencinin anormal görünümüdür.). Diğer belirti ve semptomlar şunları içerir: görme azalması, göz ağrısı ve kızarıklık ve gelişimsel gecikme. Retinoblastomlu bazı çocuklarda şaşılık (yanlış hizalanmış gözler) gelişebilir; diğer durumlarda, bir süre sonra gözün büyümesine (buftalmo) neden olabilen neovasküler glokom bulmak mümkündür.
Kanser hücreleri gözü ve diğer yapıları daha fazla istila edebilir:
- Göz içi retinoblastom. Retinoblastom, tümör tamamen gözün içinde yer aldığında göz içi olarak tanımlanabilir. Neoplazm sadece retinada bulunabilir veya koroid, siliyer cisim ve optik sinirin bir kısmı gibi diğer kısımları da etkileyebilir. Bu nedenle göz içi retinoblastom, gözün dışındaki dokulara yayılmaz.
- Ekstraoküler retinoblastom.Tümör, göz çevresindeki dokuları etkilemek için çoğalabilir (yörünge retinoblastom). Kanser ayrıca beyin, omurga, kemik iliği ve lenf düğümleri gibi vücudun diğer bölgelerine de yayılabilir.metastatik retinoblastom).
Orbita yayılımının varlığı, uveal tutulum ve optik sinirin invazyonu metastatik retinoblastom gelişimi için bilinen risk faktörleridir.
Teşhis
Aile öyküsünün pozitif olması durumunda kanser taraması için hasta düzenli olarak göz muayenesinden geçmektedir. Konjenital retinoblastom iki taraflı ise, genellikle yaşamın ilk yılında teşhis edilirken, sadece bir gözü etkilediğinde, tümörün varlığı yaklaşık 18-30 aylıkken doğrulanabilir.
Retinoblastomun klinik tanısı, fundus muayenesi ile konur.Konuma bağlı olarak tümör, dolaylı oftalmoskopi yoluyla gözün basit bir muayenesi sırasında görülebilir. Tanıyı doğrulamak, tümörün evresini (nerede olduğu, ne kadar yaygın olduğu, vücuttaki diğer organların işlevlerini etkileyip etkilemediği vb.) tanımlamak ve tedavinin etkili olup olmadığını belirlemek için görüntüleme teknikleri kullanılabilir. . Araştırmalar ultrason, bilgisayarlı tomografi (BT) ve manyetik rezonans görüntülemeyi (MRI) içerebilir.
Moleküler-genetik tanı, RB1 geninin mutasyonunun tanımlanmasıyla mümkündür.Periferik kan lenfositlerinin sitogenetik analizi (yani kromozomların), kromozom 13'ü içeren delesyonları veya yeniden düzenlemeleri tespit etmek için kullanılır (13q14.1-q14.2) .
Tedaviler
Retinoblastom durumunda çeşitli tedavi seçenekleri kullanılabilir.
Tedavinin amaçları şunlardır:
- Tümörü ortadan kaldırın ve hastanın hayatını kurtarın;
- Mümkünse gözü koruyun;
- Vizyonu mümkün olduğunca koruyun;
- Özellikle kalıtsal retinoblastomlu çocuklarda tedavinin de neden olabileceği diğer kanserlerin gelişmesini önleyin.
Prognoz (iyileşme olasılığı) ve tedavi seçenekleri aşağıdaki faktörlere bağlıdır:
- Tümörün evresi;
- Hastanın yaşı ve genel sağlık durumu;
- Tümör odaklarının yeri, boyutu ve sayısı;
- Kanserin göz küresi dışındaki diğer bölgelere yayılması
- Bir veya iki gözde görmenin korunabilme olasılığı ne kadardır?
Çoğu retinoblastom vakası, kanser göz küresinin dışına metastaz yapmadan önce erken teşhis edilir ve başarılı bir şekilde tedavi edilir ve bu da %90'ın üzerinde bir iyileşme oranı sağlar.